G. Fiorani, A. Perosa, M. Selva
Green upgrading of renewables via methylations and carboxymethylations with non-toxic dimethyl carbonate (DMC).
Dimethyl carbonate: a versatile reagent for a sustainable valorization of renewables
Author affiliations
*Corresponding authors
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford OX1 3TA, UK
bDepartment of Molecular Sciences and Nanosystems, Ca’ Foscari University of Venice, Via Torino 155, 30172 Venezia Mestre, Italy
E-mail:selva@unive.it

Giulia Fiorani
Postdoctoral Research Fellow presso University of Oxford
Dr. Fiorani earned her PhD in Chemical Sciences from the University of Rome “Tor Vergata” (2010) on synthesis and applications of ionic liquids. After several post-doctoral experiences (University of Padua, Italy 2010-2012, Ca’ Foscari University of Venice 2012-2013), Giulia was awarded a Marie Curie Intra-European Fellow in 2014 at ICIQ (Institute of Chemical Research of Catalonia, Tarragona, Spain) working under the supervision of Prof. Arjan W. Kleij on the preparation of cyclic organic carbonates from CO2 and terpene based oxiranes. Giulia joined the Williams group in 2016 and is working on renewable based polymers.
Abstract
Dimethyl carbonate (DMC) is an environmentally sustainable compound which can be used efficiently for the upgrading of several promising renewables including glycerol, triglycerides, fatty acids, polysaccharides, sugar-derived platform molecules and lignin-based phenolic compounds. This review showcases a thorough overview of the main reactions where DMC acts as a methylating and/or methoxycarbonylating agent for the transformation of small bio-based molecules as well as for the synthesis of biopolymers. All processes exemplify genuine green archetypes since they couple innocuous reactants of renewable origin with non-toxic DMC. Each section of the review provides a detailed overview on reaction conditions and scope of the investigated reactions, and discusses the rationale behind the choice of catalyst(s) and the proposed mechanisms. Criticism and comments have been put forward on the pros and cons of the described methods and their perspectives, as well as on those studies which still require follow-ups and more in-depth analyses.



Currently: Associate professor of Organic Chemistry with tenure.
Department of Molecular Sciences and Nanosystems, University Ca’ Foscari Venice.
Born in Venice in 1965. Married to Paola, two children: Alberto (2000) and Marta (2002).
– 2011, was offered the senior position as Associate professor of Chemistry with Tenure at UMAss Boston.
– 2005-2014 Assistant professor of Organic Chemistry with tenure (SSD CHIM/06), University Ca’ Foscari Venice.
– 2007 Visiting scientist, University of Sydney.
– 1996-2005 Post-doctoral researcher University Ca’ Foscari Venice.
– 1996 Ph.D. in Chemistry, Case Western Reserve University, Cleveland OH, USA.
– 1992 Laurea in Industrial Chemistry @ University Ca’ Foscari Venice.
– 2007 Endeavour Research Fellow (Austrlian Government, Department of Education, Employment and Workplace Relations) at the University of Sydney.
– 1992-1996 Fulbright Fellow (U.S. Department of State, International Educational Exchange Program) at Case Western Reserve University.
– 1993 CNR Research Fellow (1993) at Case Western Reserve University, Cleveland OH, USA.
– Ca’ Foscari Research Prize (2014, category Advanced Research).
– Royal Society of Chemistry International Journal Grants Awards (2007, 2009).
– CNR prize for research (1994).
– Outstanding teaching award CWRU (1993).
– Prize for the Laurea thesis from the Consorzio Venezia Ricerche (1992).
- Editorial Board memberships
– Advisory Board of the journal “Green Chemistry” (Royal Society of Chemistry, UK).
– Editorial Advisory Board of the journal “ACS Sustainable Chemistry and Engineering” (American Chemical Society, USA).
- Training and editorial activities.
– Scientific coordinator and organizer of the Summer School on Green Chemistry from 1998 to 2006 (funded by the European Commission, UNESCO, and NATO).
– Editor of the volume “Methods and Reagents for Green Chemistry” Wiley Interscience 2007.
– Editor of “Green Nanoscience”, volume 8 of the 12 volume set of the “Handbook of Green Chemistry” P. Anastas Ed., Wiley-VCH 2011.
– Author of over 60 scientific papers and chapters and of one patent in the field of organic chsmistry, with emphasis on green chemistry. Hirsch index (Scopus, Feb. 2014) = 21.
– Green chemistry applied to the upgrading of bio-based chemicals: towards sustainable chemical production. University of Sydney, 19 March 2014.
– Sustainable (Chemical) Solutions, Rethinking Nature in Contemporary Japan, Università Ca’ Foscari, Venezia, 25-26 February 2013
– Carbonate based ionic liquids and beyond, Green Solvents Conference, Frankfurt am Main, Dechema Gesellschaft fur Chemische Technik und Biotechnologie e. V., pp. 27, Green Solvents for Synthesis, Boppard, 8-10 Ottobre 2012
– Chemicals e Fuels da Fonti Rinnovabili, Bioforum. Biotecnologie: dove scienza e impresa si incontrano, Milano, ITER, vol. VII Edizione, Bioforum, Confindustria Venezia, 24.02.2011
– Green Chemistry for Sustainability: Teaching ionic liquids new tricks & A breath of oxygen for bio-based chemicals., Slovenian-Italian conference on Materials and Technologies for Sustainable Growth, Ajdovscina, Slovenia, 4-6 Maggio 2011
– Benign molecular design, WORKSHOP ON ECOPHARMACOVIGILANCE, Verona, 26-27 Marzo 2009
– Not merely solvents: task specific ionic liquids made by green syntheses, COIL-3 Pre-symposium workshop, Cairns, Australia, 31/05/2009
– Multiphase catalysis: a tool for green organic synthesis, Royal Australian Chemical Institute NSW Organic Chemistry Group, 28th Annual One-Day Symposium, MacQuarie University, Sydney, Australia, 5 December 2007
– Catalytic Reactions in Liquid Multiphasic Systems The acronym talk, INTAS Project on POPs, Moscow, 12-14 Giugno 2005
– Catalytic reactions in liquid multiphasic systems, Convegno: Eurogreenpol – First European Summer School on Green Chemistry of Polymers, Iasi – Rumania, 21-27 Agosto 2005
– Multiphase hydrodehalogenation reactions, RWTH Aachen – Germany, 12 Febbraio 2003
– Mechanism and Synthetic Applications of the Multiphase Catalytic Systems, International Workshop on Hazardous Halo-Aromatic Pollutants: Detoxification and Analysis, Venezia, 14-16 Maggio 2002
– The multiphase catalytic hydrodehalogenation of haloaromatics, European Summer School on Green Chemistry, Venezia, 10-15 September 2001
– Quality assurance board of Ca’ Foscari University
– Teaching council of the International College, Ca’ Foscari merit school.
– Academic Council of Venice International University VIU.
– Delegate for international relations of the Department of Molecular Sciences and Nanosystems.
– Scientific board of Edizioni Ca’ Foscari – Digital Publishing.
– Research committee of the Department of Molecular Sciences and Nanosystems.
– Teaching board of the Doctorate in Chemical Sciences (2012-2014).
– Teaching board of the degree course Bio- and Nanomaterials science and Technology.
– Erasmus selection committee.
– Overseas selection committee
– Post-doctoral selection committees.
- Referee, reviewer, and examiner for:
– Valutazione della Qualità della Ricerca (VQR), ANVUR
– Progetti di Rilevante Interesse Nazionale (PRIN), MIUR
– American Chemical Society Petroleum Research Fund (USA).
– Ph.D. Theses, University of Nottingham (UK) and University of Sydney (Aus).
– European Science Foundation
– Journals published by: Royal Society of Chemistry, American Chemical Society, Wiley, Elsevier, Springer, IUPAC
– Coordinator of a Cooperlink project funded by the Italian Ministry for Education, University and Research, 2011, 12 months, entitled “Joint PhD between Università Ca’ Foscari and the University of Sydney: integration of experiment and theory towards the green synthesis of self-assemblying materials and the use of renewable resources”.
– Participant in the Project of Relevant National Interest (PRIN) “Green organic syntheses mediated by new catalytic systems”, 2010, 24 months.
– Tutor of a PhD scholarship funded by the Regione Veneto through the European Social Fund, entitled “Organic syntheses of active principles and chemicals for the pharmaceutical industry using green solvents “ 2009-2011, 36 months.
– Principal Scientist of a post-doctoral fellowship funded by the Regione Veneto through the European Social Fund entitled “New reduced environmental impact chemical synthesesfor the preparation of monomers for advanced polymers, April 2012, 12 months.
– Principal Scientist of a post-doctoral fellowship funded by the Regione Veneto through the European Social Fund entitled “Environmentally compatible chemical syntheses of fluorinated monomers for advanced materials” April 2013, 12 months.
– Principal Scientist of a post-doctoral fellowship funded by the Regione Veneto through the European Social Fund entitled “Valorisation of renewable substrates from biomass, such as glycerol and its derivatives, using green chemistry” April 2014, 12 Months
– Principal Scientist of a research contract between the chemical company Aussachem (Santandrà di Povegliano, TV), entitled: “Green Chemistry for the valorisation of glycerol and of its derivatives: new ecofriendly products” December 2013.
- International collaborations and networks
– Teaching and research collaboration with the University of Sydney, School of Chemistry Laboratory for Advanced Catalysis and Sustainability prof. Thomas Maschmeyer. A joint PhD program in Chemistry was established and is currently running. Up to date 5 students (3 outgoing, 2 incoming) have benefited from this agreement The first joint PhD has been awarded in December 2013 (Marina Gottardo). Four joint publications have already been produced, and others are in preparation.
– Research collaboration with the Queen’s University of Belfast, Queen’s University Ionic Liquids Laboratory, prof. Kenneth R. Seddon, for the exchange of Erasmus students who carry out research towards their MS thesis. Currently the student Riccardo Zabeo is in Belfast w research towards his thesis, tutor dr. Perosa. Previously, the PhD student Marco Noè (tutor Perosa) spent 4 months in Belfast carrying out research that was published on an international journal.
– In the framework of a scientific collaboration with prof. Janet Scott of the Centre for Sustainable Chemical Technologies of the University of Bath, an Erasmus Mundus Joint Doctorate project entitled “Bio-Based Chemicals and Materials” was submitted in 2011 and was evaluated positively albeit not funded. Nonetheless the collaboration has already produced a joint publication.
– Summer School on Green Chemistry Network. Following the 8 editions of the “Summer school on Green Chemistry” (1998-2005) coordinated and organized by the applicant, a Green Chemistry Network was initiated that involves the following institutions: RWTH-Aachen, QUB-QUILL Belfast, UNSW-Sydney, ARKEMA-France, University of Groningen-NL, Dow Europe-CH, Universite de Poitiers, ETH-Zurich, TU-Darmstadt, Universidad Politecnica de Valencia, Delft University of Technology, TU-Munchen.
– Since 1993 Alvise Perosa is a member of the American Chemical Society.
- MoU’s and International agreements
– Alvise Perosa started the Joint PhD degree in Chemistry between the University of Sydney and the Università Ca’ Foscari Venezia.
– Erasmus, Alvise Perosa is the contact person for the following Erasmus agreements: Universitat Autonoma de Barcelona, Universidad Rey Juan Carlos, Universidad Rovira i Virgili,UNIVERSITE D’AVIGNON ET DES PAYS DE VAUCLUSE, ARISTOTLE UNIVERSITY THESSALONIKI, Queen’s University of Belfast.
– Marco Noè (PhD 2009-11: 24° cycle)
– Jessica N. G. Stanley (PhD cotutelle University of Sydney, 2012-2014)
– Alessio Caretto (PhD 2012-14: 27° cycle)
– Manuela Facchin (PhD 2014-16: 29° cycle)
– Tutor if BSc and MSc level students of the degree corse in Sustainable Chemistry and Technologies and, and of the MSc degree course in Science and Technolgy of Bio- and Nanomaterials.
– 1992-94, Case Western Reserve University, Chemistry BS: Organic Chemistry 1 Laboratory (teaching assistant award in 1993).
– 1997-2000, Università Ca’ Foscari Venezia, degree course in Environmental Sciences: Organic Chemistry Exercises.
– 1997-2000, Università Ca’ Foscari Venezia, degree course in Industrial Chemistry: Organic Chemistry 1 & 2 Laboratory, Industrial Chemistry 2 Exercises, Organic Chemistry 1 (part-time students) and Advanced Organic Chemistry.
– 2006-09, Università Ca’ Foscari Venezia, degree course in Chemical Sciences and Technologies for Cultural Heritage Conservation and Restoration: Organic Chemistry Laboratory.
– 2006-07, Università Ca’ Foscari Venezia, degree course in Chemistry, Industrial Chemistry, Materials Chemistry, Environmental Sciences: Organic Chemistry 1 and Laboratory for part-time students.
– 2005-06, 2011-12, 2012-13, 2013-14: Università Ca’ Foscari Venezia, degree course in Chemistry and in sustainable Chemical Technologies: Organic Chemistry 2 and Laboratory.
– 2011-12, Università Ca’ Foscari Venezia, degree course in Chemistry and in sustainable Chemical Technologies: Green Organic synthesis Laboratory.
– 2012-13, 2013-14 Università Ca’ Foscari Venezia, MS degree course in Bio e Nanomaterials: Colloids and Interfaces.
– 2013-14 Università Ca’ Foscari Venezia, Graduate course in Organic syntheses from renewable building blocks.










 Paul T. Anastas,
Paul T. Anastas,

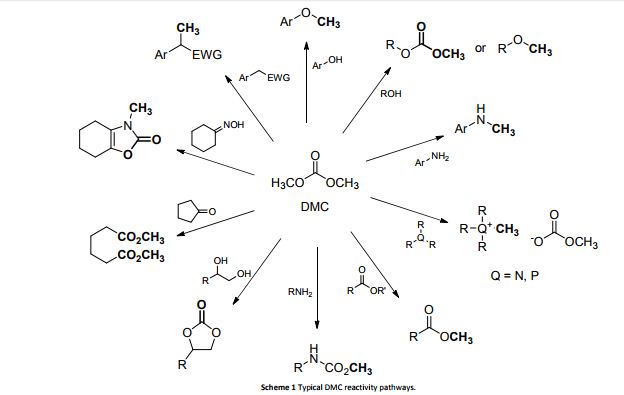
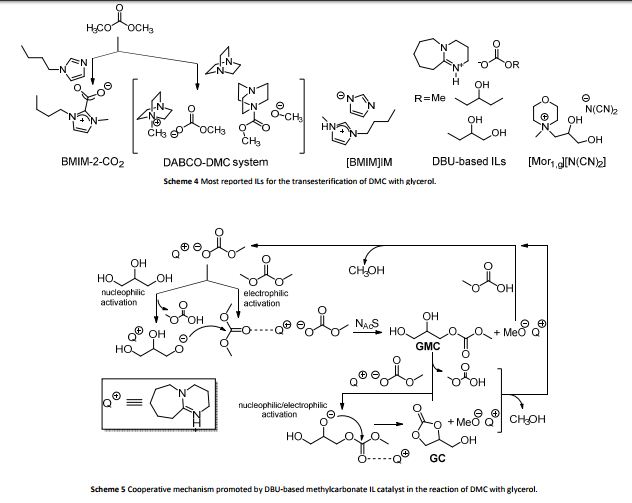



















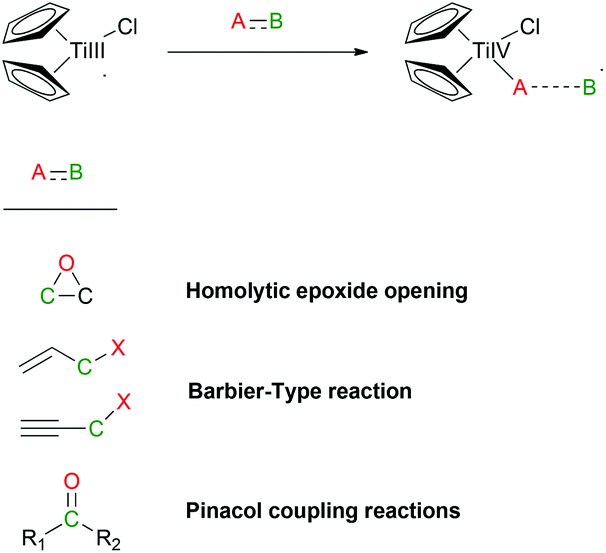

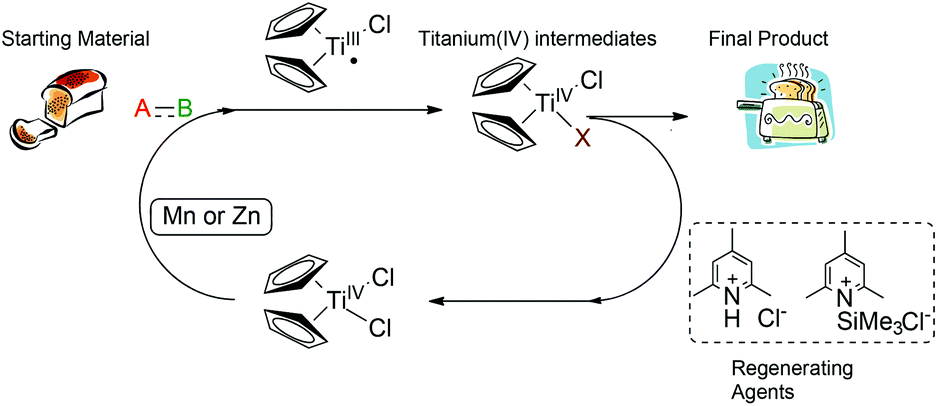









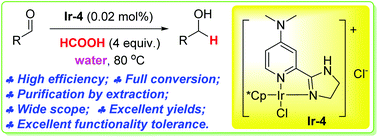
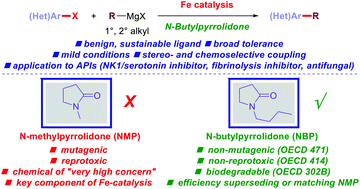
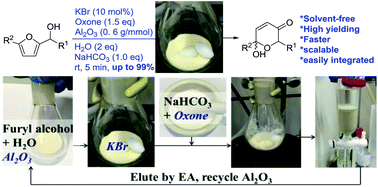
 800 mol mol−1 h−1. A wide variety of functional groups, such as electron-rich or deficient (hetero)arenes and alkenes, alkyloxy groups, halogens, phenols, ketones, esters, carboxylic acids, cyano, and nitro groups, are all well tolerated, indicating excellent chemoselectivity.
800 mol mol−1 h−1. A wide variety of functional groups, such as electron-rich or deficient (hetero)arenes and alkenes, alkyloxy groups, halogens, phenols, ketones, esters, carboxylic acids, cyano, and nitro groups, are all well tolerated, indicating excellent chemoselectivity.








_reduced.jpg)





